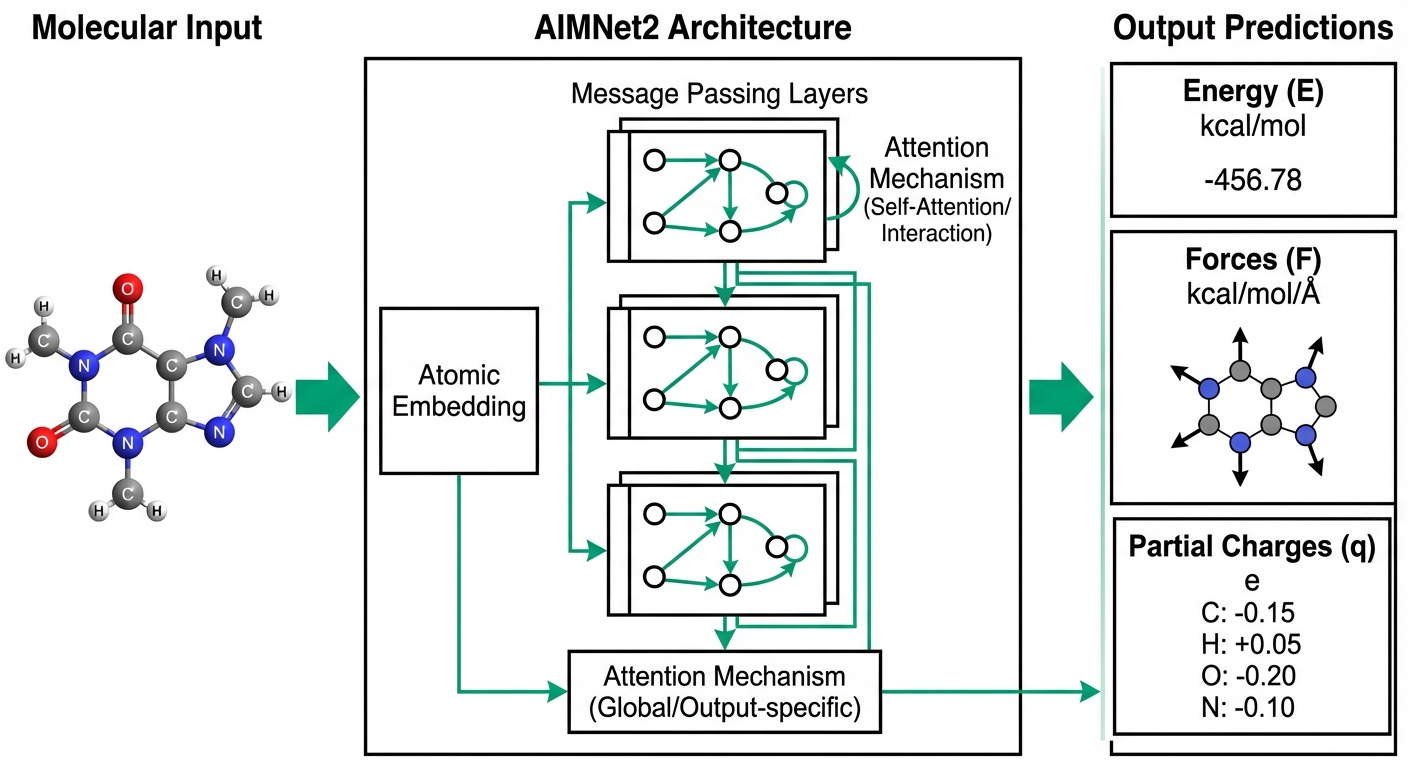
Machine Learning Potentials
Transferable neural network potentials bridging quantum accuracy with computational efficiency
Explore
My research focuses on developing machine learning methods for molecular sciences, with applications ranging from drug discovery to materials design. The work spans quantum chemistry, neural network potentials, generative models, and high-throughput computational screening.
Transferable neural network potentials bridging quantum accuracy with computational efficiency
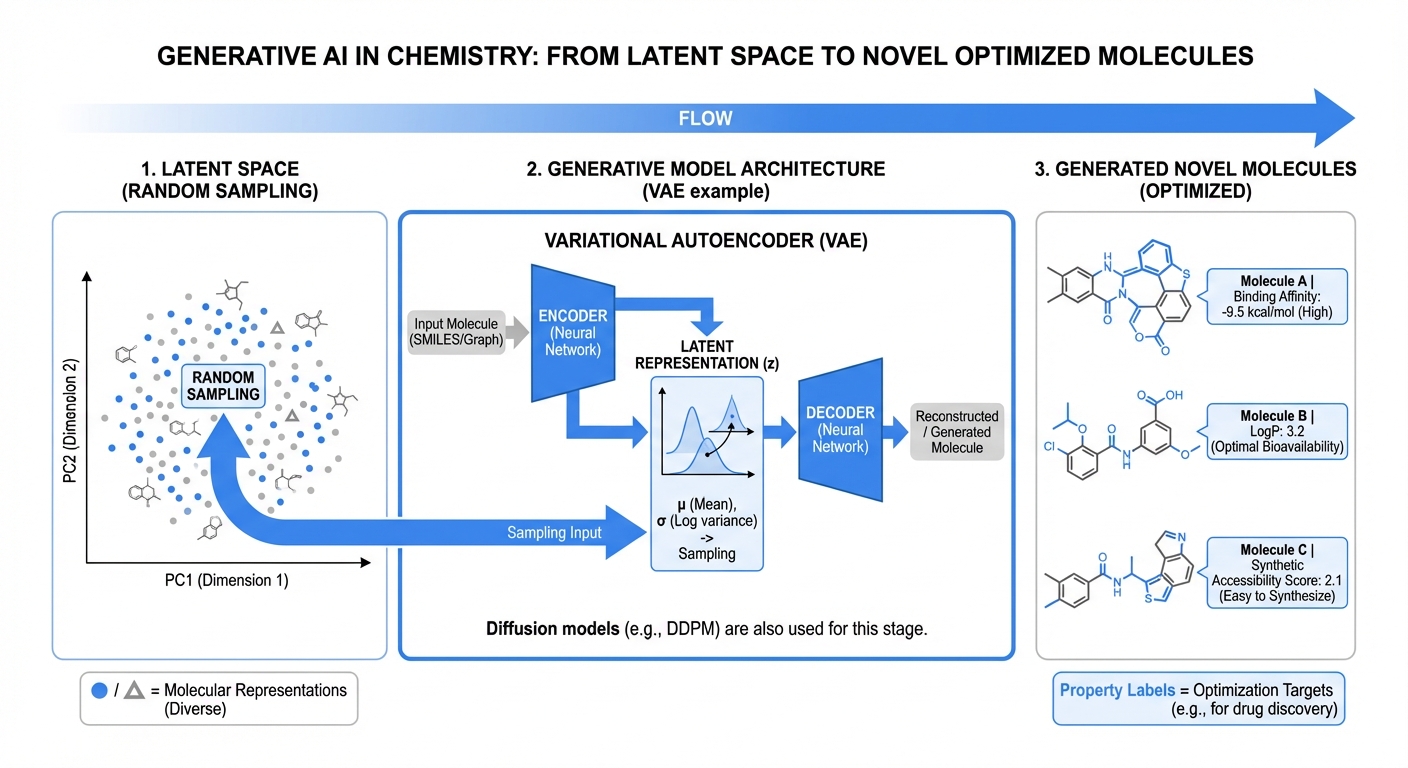
Generative models enabling systematic exploration of chemical space under biological and synthetic constraints
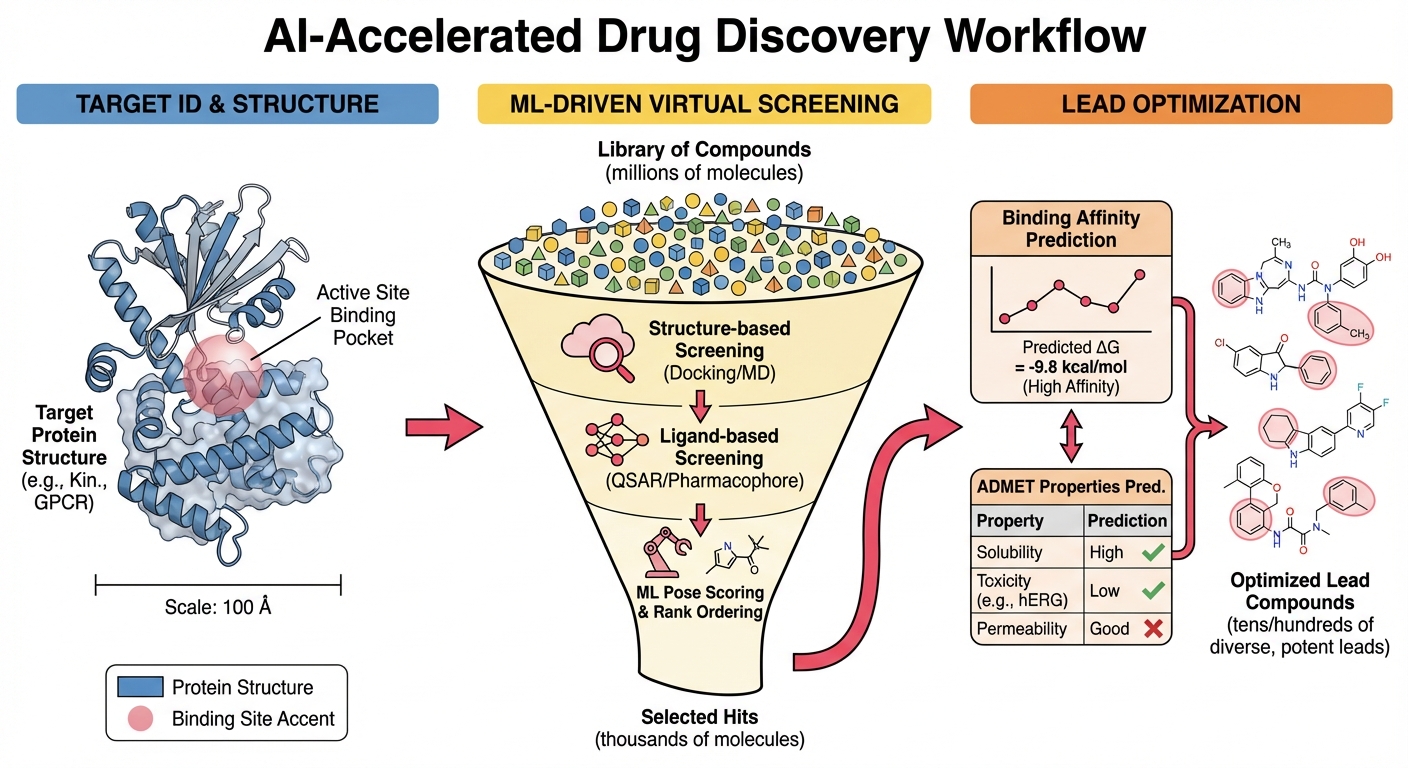
Energy-based ML methods for reliable compound prioritization in industrial drug discovery
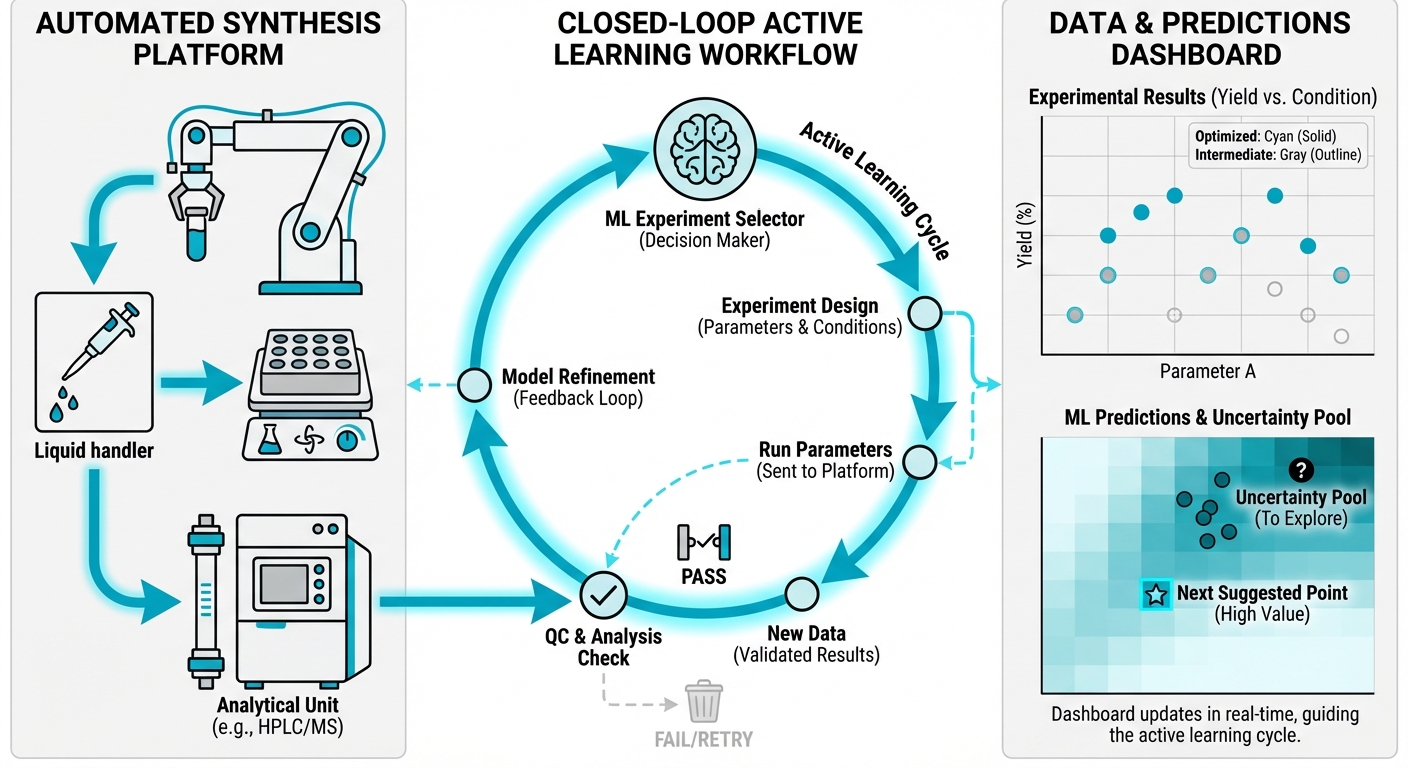
ML-enabled workflows for reproducible chemical experimentation in cloud laboratories
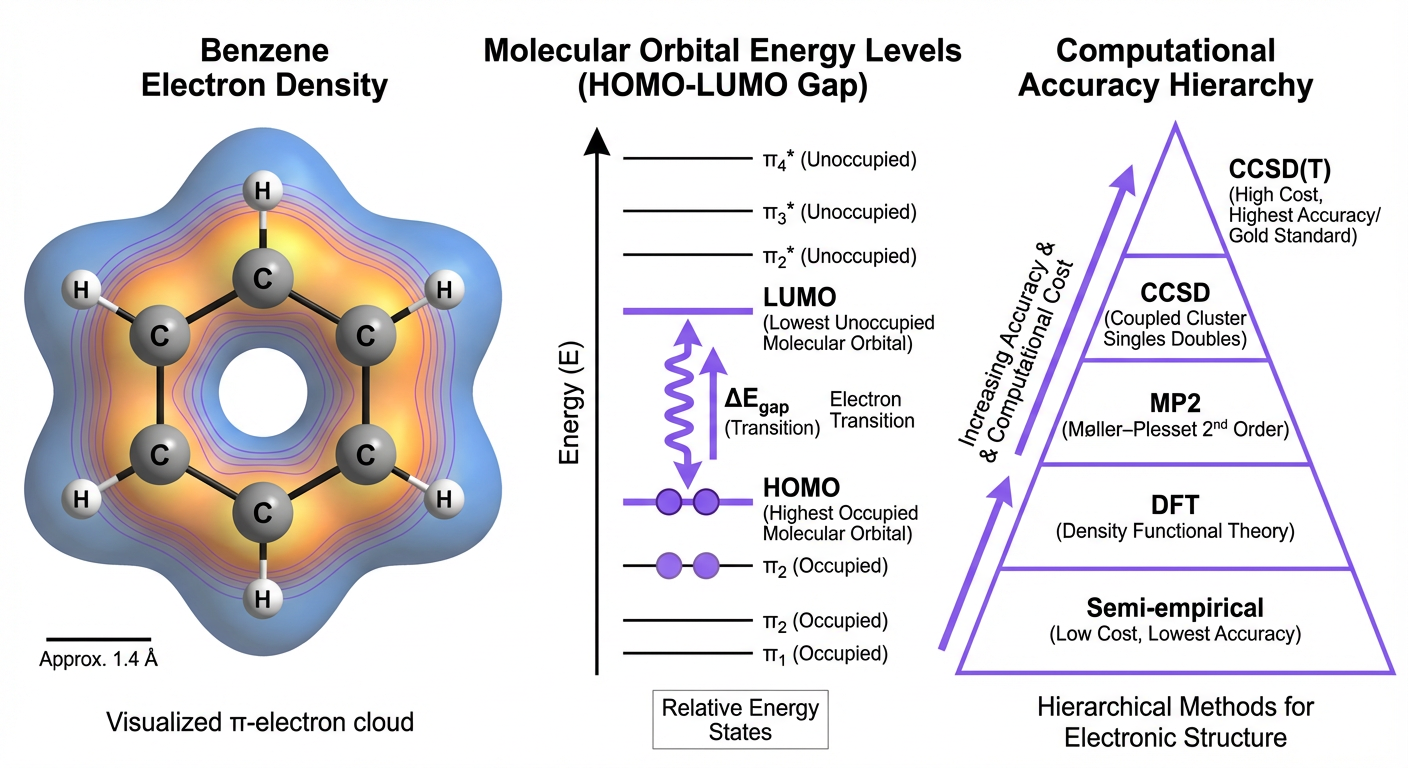
First-principles electronic structure methods providing the theoretical foundation for ML potential development
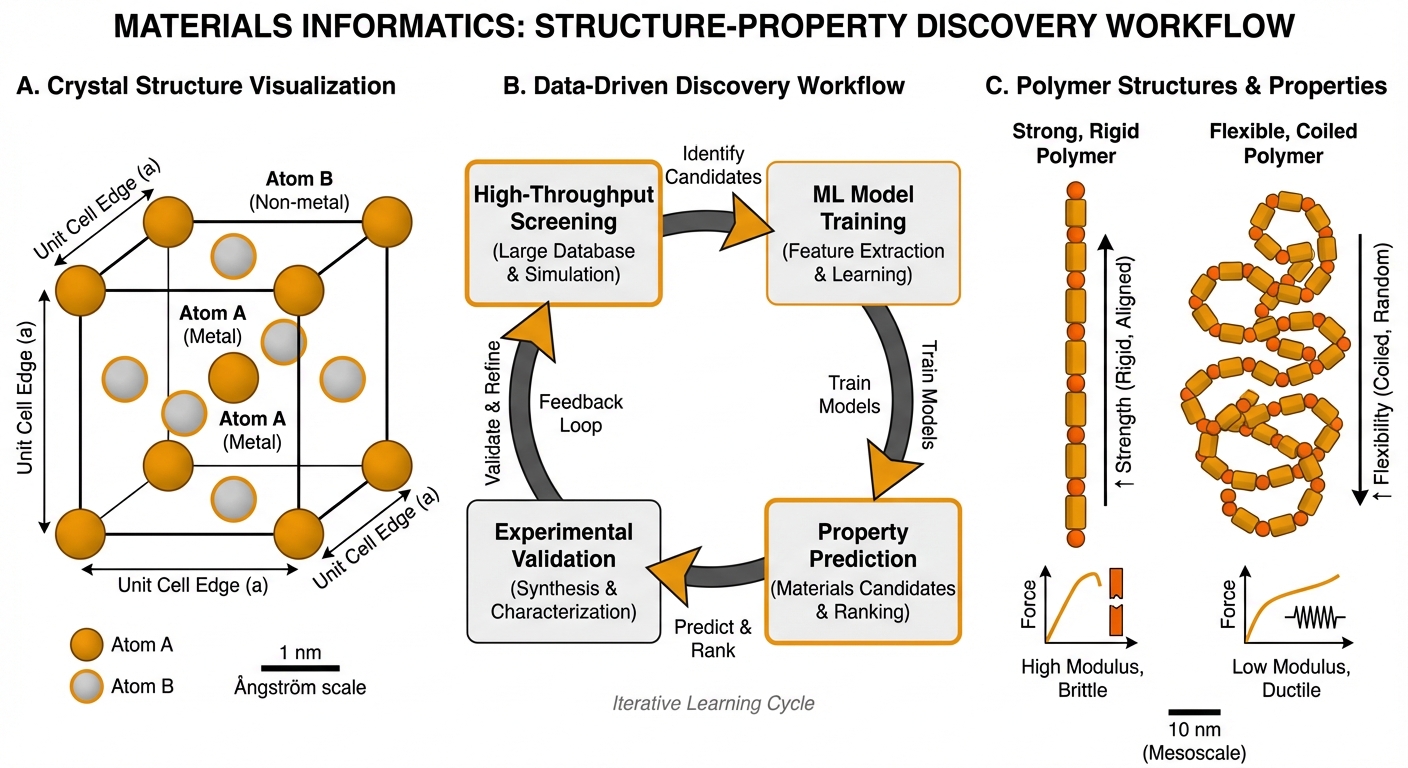
Machine learning-accelerated discovery and optimization of advanced functional materials

ML-accelerated reaction modeling enabling exploration of chemical transformations at unprecedented scale
We believe that machine learning will fundamentally transform how chemistry is practiced. Our goal is to develop methods that are not just accurate, but also interpretable, transferable, and practically useful for real-world chemical problems.
Our approach combines rigorous physical foundations with modern deep learning techniques. By encoding physical laws and chemical knowledge into neural network architectures, we create models that are both data-efficient and physically meaningful.
We actively collaborate with experimental chemists, pharmaceutical companies, and national laboratories. Our open-source tools are used by researchers worldwide and have been integrated into several commercial platforms.
If you're interested in collaboration opportunities, please visit the contact page for more information.