Data-Driven Discovery of Functional Materials
Materials Informatics
Applying machine learning and computational methods to accelerate the discovery and optimization of functional materials, from polymers to crystalline solids.
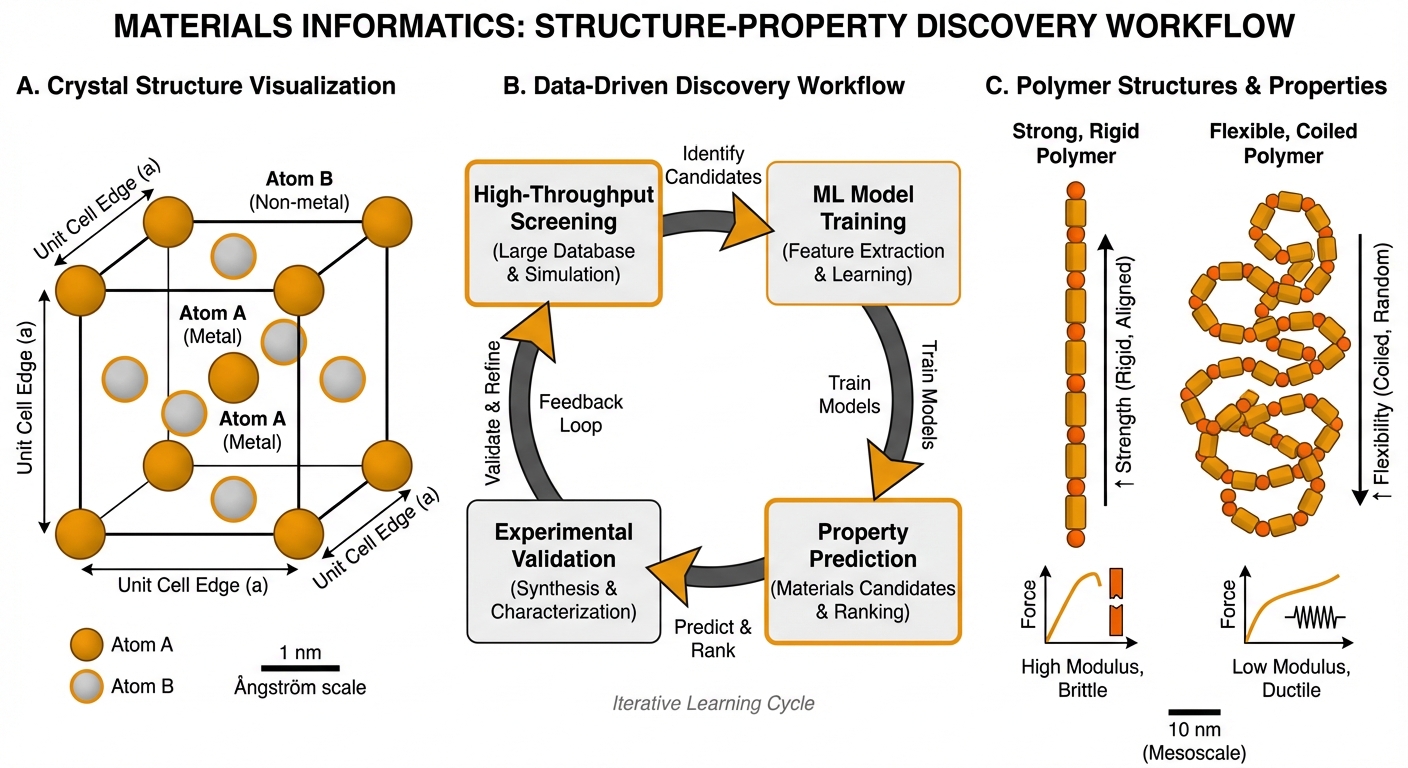
Materials informatics represents the application of data science, machine learning, and computational methods to accelerate the discovery and optimization of functional materials. My research in this area bridges molecular-scale modeling—where machine learning potentials provide quantum-level accuracy—with materials-scale design challenges that require navigating vast compositional and processing spaces. The goal is to replace traditional trial-and-error materials development with data-driven approaches that systematically explore structure-property relationships.
Polymer Design and Optimization
A significant focus of my materials informatics research involves polymeric materials, where the complexity of molecular weight distributions, processing conditions, and multi-component formulations creates design spaces that are intractable for exhaustive experimental exploration. Machine learning approaches enable efficient navigation of these spaces by learning structure-property mappings from experimental or computational data.
Recent work on 3D-printable elastomers demonstrates the power of human-in-the-loop reinforcement learning for materials optimization. Starting with a training set of 92 polyurethane formulations, we developed a multi-component reward system that guides reinforcement learning agents toward materials exhibiting both high strength and high extensibility—properties that typically trade off against each other. Through iterative optimization combining ML predictions with expert chemical intuition, we discovered elastomers with more than double the average toughness compared to the initial dataset. The exploitation phase identified twelve materials achieving both high strength (greater than 10 MPa) and high strain at break (greater than 200%), outcomes that substantially exceed what conventional optimization would achieve.
This work illustrates several principles central to my approach: combining data-driven methods with domain expertise rather than treating ML as a black box; explicitly addressing multi-objective trade-offs that characterize real materials design problems; and maintaining human oversight while leveraging computational efficiency for exploration.
Crystal Structure Prediction
Crystalline materials present unique challenges for computational discovery, as the arrangement of molecules in the solid state profoundly influences properties ranging from mechanical behavior to pharmaceutical bioavailability. Crystal structure prediction (CSP) aims to identify thermodynamically stable and kinetically accessible polymorphs from molecular structure alone—a problem that has been called one of the grand challenges of computational chemistry.
My research applies machine learning potentials to enable efficient and accurate CSP at scale. The AIMNet2 framework provides the energy evaluations needed for geometry optimization and polymorph ranking at costs that enable exploration of thousands of candidate structures. Recent work on celecoxib—a widely used COX-2 inhibitor—demonstrates this capability: GPU-accelerated crystal structure generation combined with quasi-harmonic free-energy corrections successfully reproduced the experimental energy hierarchy of known polymorphs while identifying novel low-energy structures with distinct packing motifs. Beyond thermodynamic ranking, we evaluated elastic properties and thermal expansion effects across polymorphs, revealing structural features that underpin mechanical flexibility and stability.
This work has direct pharmaceutical relevance, where polymorph control is essential for intellectual property, manufacturing consistency, and drug performance. It also demonstrates the broader applicability of ML potentials to periodic systems and solid-state chemistry.
Property Prediction and QSPR
Quantitative structure-property relationships (QSPR) remain a cornerstone of materials informatics, enabling rapid property estimation from molecular or compositional descriptors. My research has developed and applied QSPR approaches for diverse materials properties including electronic properties such as bandgaps, dielectric constants, and charge mobility; thermal and mechanical properties relevant to engineering applications; stability and degradation behavior under operational conditions; and toxicity and environmental impact for sustainable materials design.
The integration of physics-based descriptors with machine learning models often outperforms purely data-driven approaches, particularly in extrapolative regimes where training data is sparse. Descriptors derived from quantum chemical calculations—orbital energies, electron densities, electrostatic potentials—encode physical information that statistical models can leverage for improved generalization.
High-Throughput Screening
Materials discovery increasingly relies on high-throughput computational screening to identify promising candidates before experimental synthesis. My research contributes to this paradigm through efficient property prediction using ML potentials and surrogate models, systematic exploration of compositional and structural variation, uncertainty quantification to prioritize experimental validation, and integration with experimental workflows for closed-loop optimization.
The combination of ML potentials with automated experimentation—discussed in my work on experiment automation—creates opportunities for materials discovery cycles that iterate between computational prediction and experimental validation with minimal human intervention.
Outlook
Materials informatics is evolving toward increasingly integrated and autonomous discovery platforms. Key directions for my research include extending ML potentials to broader classes of inorganic and hybrid materials; developing generative models for materials composition and structure; integrating computational screening with robotic synthesis and characterization; and advancing toward self-driving laboratories that autonomously navigate materials design spaces.
The long-term vision is to transform materials development from an empirical art into a systematic science, where computational methods guide experimental effort toward the most promising regions of materials space while continuously learning from outcomes to improve future predictions.