Energy-Driven Computational Methods for Medicinal Chemistry
AI for Drug Discovery
Developing energy-driven, decision-oriented computational methods that integrate ML potentials, free-energy simulations, and active learning for drug discovery.
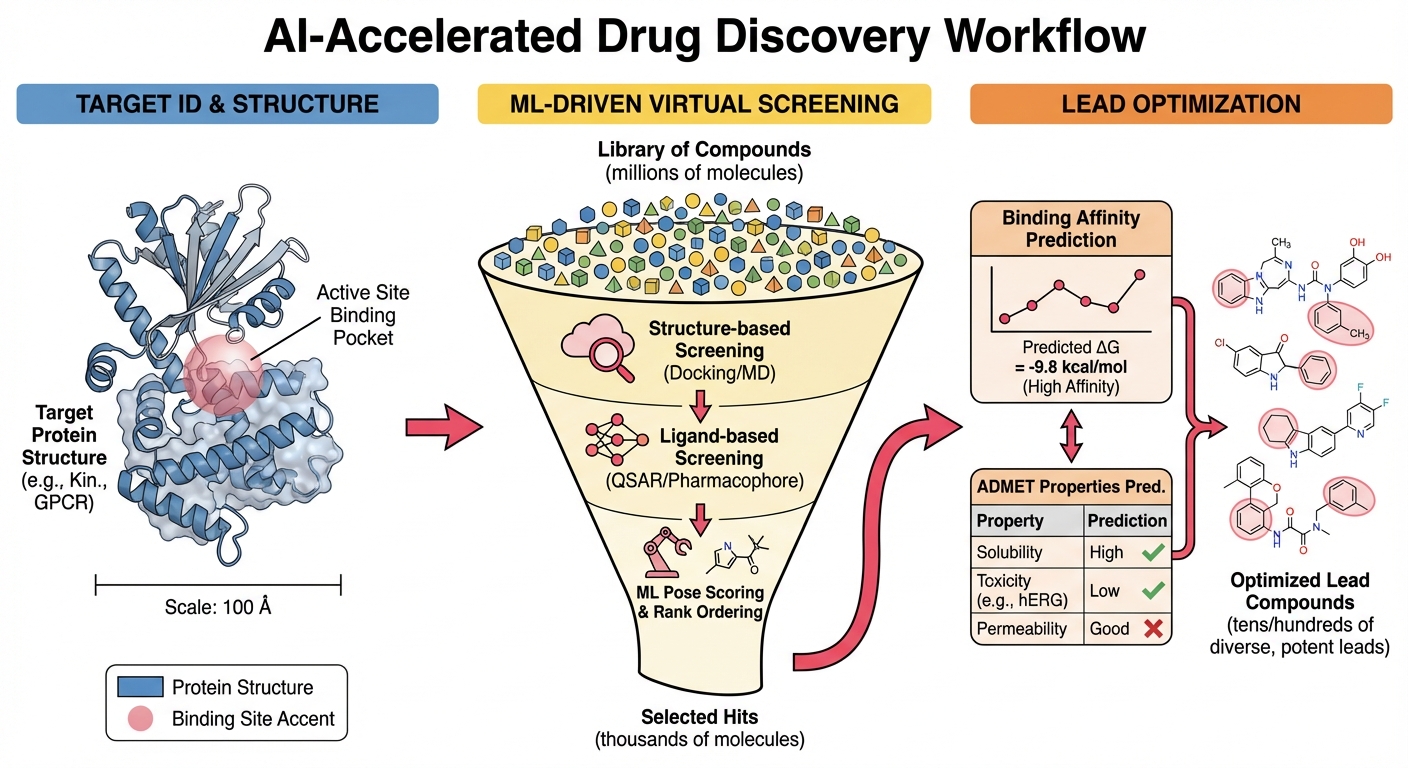
My research in drug discovery focuses on developing energy-driven, decision-oriented computational methods that support medicinal chemistry and early discovery under industrial constraints. Rather than emphasizing molecule generation in isolation, this work targets the reliable prioritization of compounds by integrating machine learning interatomic potentials, free-energy simulations, reaction-aware feasibility assessment, and active learning into unified workflows. This approach is motivated by a central challenge in pharmaceutical development: reducing cycle time and risk when making decisions under uncertainty, especially for novel chemotypes and chemically complex lead series where historical data provides limited guidance.
Energy-Based Modeling for Medicinal Chemistry
A core component of my research program is the application of transferable machine learning potentials to enable accurate energetic evaluation of drug-like molecules at scale. The AIMNet2 framework provides near-quantum chemical accuracy for neutral molecules, ions, radicals, and reactive intermediates while maintaining computational costs that enable high-throughput deployment. This combination makes ML potentials particularly well-suited for medicinal chemistry applications where conformational strain, intramolecular interactions, and protonation states strongly influence both binding affinity and developability properties.
The AIMNet2-NSE extension enables treatment of open-shell and spin-polarized systems, which arise frequently in medicinal chemistry contexts including redox-active compounds such as quinones and flavins, covalent inhibitors that form radical intermediates during target engagement, and metabolically relevant species generated during cytochrome P450-mediated oxidation. Together, these models provide a physically consistent foundation for downstream sampling, ranking, and optimization in drug discovery campaigns, complementing rather than replacing established medicinal chemistry intuition with quantitative energetic assessments.
Applications of this framework extend to conformational strain analysis for ligand binding, crystal structure prediction relevant to solid-state formulation, and ring strain energy quantification for strain-promoted reactions increasingly used in bioconjugation chemistry. The pKa-ANI model, published in Chemical Science, represents the first empirical ML approach that significantly outperforms null models for all titratable protein residues, enabling rapid assessment of pH-dependent binding interactions and ADME properties that depend on molecular ionization state.
ML-Accelerated Free Energy Simulations
Reliable prioritization in drug discovery increasingly depends on free-energy-based methods, particularly during lead optimization where small structural modifications must be rank-ordered with confidence. A key limitation of conventional workflows is the computational cost of force evaluations in molecular dynamics simulations, which restricts both sampling depth and compound throughput.
By accelerating energy and force calculations, transferable ML potentials enable high-throughput free energy simulations encompassing conformational free energies that quantify ligand reorganization penalties, relative binding free energies for congeneric series comparison, and solvation free energies relevant to solubility and membrane permeability. Collaborative work has demonstrated that ANI2x/AMBER14SB hybrid potentials reduce errors in absolute binding free energies from 0.97 kcal/mol with pure molecular mechanics to 0.47 kcal/mol with ML/MM hybrid approaches—approaching the chemical accuracy threshold critical for reliable rank-ordering in medicinal chemistry campaigns.
My group’s active learning-guided lead optimization methodology, published in the Journal of Chemical Information and Modeling, demonstrates that combining thermodynamic integration with AutoML and active learning can achieve 20-fold efficiency gains compared to brute-force screening while maintaining prediction accuracy within 1-2 kcal/mol of experimental values. Application to SARS-CoV-2 papain-like protease identified 133 compounds with improved binding affinity from thousands of candidates, with 16 compounds showing greater than 100-fold affinity improvement—outcomes that substantially exceed typical expert-guided medicinal chemistry campaigns.
Reaction Awareness and Chemical Feasibility
Drug discovery is inseparable from chemical feasibility considerations. Synthesis routes, late-stage functionalization options, and metabolic transformations all impose practical constraints that must be considered early to reduce downstream attrition. Compounds that are computationally attractive but synthetically intractable or metabolically unstable represent wasted optimization effort.
My research addresses this dimension through large-scale reaction modeling that enables systematic exploration of chemical transformations alongside molecular optimization. The AIMNet2-rxn framework enables efficient generation and evaluation of millions of reaction pathways, supporting synthesis-aware discovery that extends beyond curated reaction template databases. Related work demonstrates the applicability of transferable ML potentials to catalytic and organometallic systems central to pharmaceutical synthesis, including cross-coupling chemistry and C-H activation reactions commonly employed in late-stage diversification.
This reaction-aware perspective enables energetic cost assessment during lead optimization by quantifying whether proposed modifications introduce unfavorable strain or electronic effects, feasibility filtering early in the design process before synthesis resources are committed, synthesis route prediction integrated with molecular design rather than treated as a separate downstream problem, and metabolic stability estimation through identification of sites susceptible to oxidative metabolism based on reaction energetics.
Active Learning and Adaptive Decision-Making
A defining feature of my approach to computational drug discovery is the use of active learning to guide both computational simulation and experimental validation. Rather than exhaustively screening chemical space—an approach that becomes intractable as compound libraries grow—models identify regions of high uncertainty or high expected value and selectively request additional data in those areas.
This paradigm aligns naturally with industrial constraints where computational budgets, assay capacity, and synthesis throughput are all limited. Active learning enables rapid model improvement with minimal data acquisition, focus on decisions with the greatest expected impact on project outcomes, efficient utilization of precious synthesis and assay resources, and elimination of redundant calculations and experiments that provide diminishing information returns.
The integration of active learning with ML potentials and free energy methods creates a principled framework for compound prioritization under uncertainty—acknowledging that models are imperfect while still extracting maximum value from their predictions.
CACHE and Community Benchmarks
My research aligns closely with the goals of CACHE (Critical Assessment of Computational Hit-finding Experiments), a public-private partnership that emphasizes prospective evaluation, reproducibility, and decision relevance under realistic discovery constraints. Unlike retrospective benchmarks that can be subject to data leakage and overfitting, CACHE involves blind predictions on undisclosed targets followed by experimental synthesis and testing of computational nominations.
In the inaugural CACHE Challenge targeting the LRRK2 WD40 domain—a Parkinson’s disease target with no previously known small-molecule inhibitors—my team achieved a tied first-place finish. The methodology combined Deep Docking across 4.1 billion molecules from the Enamine REAL library with absolute binding free energy calculations via molecular dynamics-based thermodynamic integration, achieving an 8.5% experimental hit rate that substantially exceeds typical virtual screening outcomes.
Energy-based ML models, free-energy simulations, and active learning workflows provide a natural foundation for addressing CACHE-style challenges that demand robust ranking under limited data, principled uncertainty quantification for decision support, generalization beyond historical datasets to novel chemical space, and reproducible transparent methods that can be independently evaluated. This work contributes toward next-generation benchmarking paradigms that reflect how computational methods are actually deployed in industrial drug discovery, providing more meaningful assessments than retrospective metrics alone.
Outlook
Looking forward, my research aims to further integrate ML-accelerated free-energy methods, reaction-aware modeling, and active learning into end-to-end discovery platforms. The long-term objective is to provide decision-focused computational tools that reduce cycle time in lead optimization by enabling faster iteration between design hypotheses and experimental validation, improve prioritization accuracy through physics-based assessment rather than purely statistical correlations, enable reliable exploration of novel chemical space where historical data is sparse or absent, and support industrial drug discovery under real-world resource constraints.
By combining physics-based rigor with data-driven efficiency, these methods aim to accelerate the translation of computational predictions into therapeutic candidates—not by replacing medicinal chemistry expertise, but by augmenting it with quantitative tools for evaluating molecular properties that are difficult to assess through intuition alone.
Key Publications
Active Learning Guided Drug Design Lead Optimization Based on Relative Binding Free Energy Modeling
Journal of Chemical Information and Modeling , 63 , 583–594 (2023)
The transformational role of GPU computing and deep learning in drug discovery
Nature Machine Intelligence , 4 , 211–221 (2022)
A critical overview of computational approaches employed for COVID-19 drug discovery
Chemical Society Reviews , 50 , 9121–9151 (2021)
Software & Tools
Frequently Asked Questions
How do machine learning potentials improve drug discovery compared to traditional methods?
Machine learning potentials like AIMNet2 provide near-quantum chemical accuracy at computational costs that enable high-throughput deployment. This allows accurate evaluation of conformational strain, intramolecular interactions, and protonation states that strongly influence binding affinity and developability properties—analyses that would be prohibitively expensive with traditional quantum chemistry methods.
What is the CACHE challenge and how does it validate computational drug discovery methods?
CACHE (Critical Assessment of Computational Hit-finding Experiments) is a public-private partnership that uses prospective evaluation with blind predictions on undisclosed targets followed by experimental synthesis and testing. In the inaugural CACHE Challenge targeting the LRRK2 WD40 domain for Parkinson's disease, our team achieved a tied first-place finish with an 8.5% experimental hit rate, substantially exceeding typical virtual screening outcomes.
How does active learning reduce computational costs in drug discovery?
Active learning-guided lead optimization combines thermodynamic integration with AutoML to achieve 20-fold efficiency gains compared to brute-force screening while maintaining prediction accuracy within 1-2 kcal/mol of experimental values. Applied to SARS-CoV-2 papain-like protease, this approach identified 133 compounds with improved binding affinity, with 16 showing greater than 100-fold improvement.
What makes pKa-ANI different from other protein pKa prediction methods?
pKa-ANI is the first empirical ML model that significantly outperforms null models for all titratable protein residues (Asp, Glu, His, Lys, Tyr), achieving mean absolute error less than 0.5 pKa units. It uses only PDB coordinates as input and runs in approximately 0.2 seconds per residue, enabling rapid assessment of pH-dependent binding interactions and ADME properties.