Learning Quantum Mechanics with Neural Networks
Machine Learning Potentials
Developing transferable ML interatomic potentials that bridge machine learning and quantum mechanics, enabling molecular simulations at unprecedented scale.
My research centers on the development of transferable machine learning interatomic potentials (MLIPs) that bridge the gap between quantum chemical accuracy and the computational efficiency required for large-scale molecular simulations. Classical force fields, while computationally tractable, remain fundamentally limited in their capacity to describe bond breaking, charge redistribution, and the full diversity of chemical environments encountered in reactive systems. First-principles electronic structure methods provide the requisite accuracy but scale unfavorably with system size, rendering extensive conformational sampling and reaction pathway exploration computationally prohibitive. Machine learning potentials offer a practical resolution to this trade-off, provided that transferability, robustness, and physical consistency are treated as primary design objectives rather than post hoc validation criteria.
Foundations in Quantum Chemistry
My early research was grounded in quantum chemistry and electronic structure theory, with applications spanning reaction mechanisms, thermochemistry, and molecular structure prediction. This work established a rigorous methodological foundation while simultaneously highlighting the central limitation of ab initio approaches: unfavorable computational scaling with system size and configurational complexity. These constraints motivated my transition toward data-driven modeling as a means to extend quantum-level accuracy to larger and more complex chemical systems. This background continues to shape my approach to machine learning, where I emphasize physically motivated architectures and chemically meaningful representations that encode known principles of molecular interactions rather than treating ML models as purely statistical tools. Understanding the trade-offs inherent to different levels of electronic structure theory—from density functional approximations to coupled-cluster methods—informs the choice of reference data, the design of training protocols, and the interpretation of model predictions in chemically relevant contexts.
Transferable Neural Network Potentials: The AIMNet Family
A central contribution of my research program is the development of the AIMNet family of neural network potentials, designed to model molecular energies and forces across broad chemical domains without retraining for each new application. The original AIMNet framework demonstrated that neural networks can achieve strong generalization when atomic environment representations respect fundamental physical constraints including locality, extensivity, and chemical similarity.
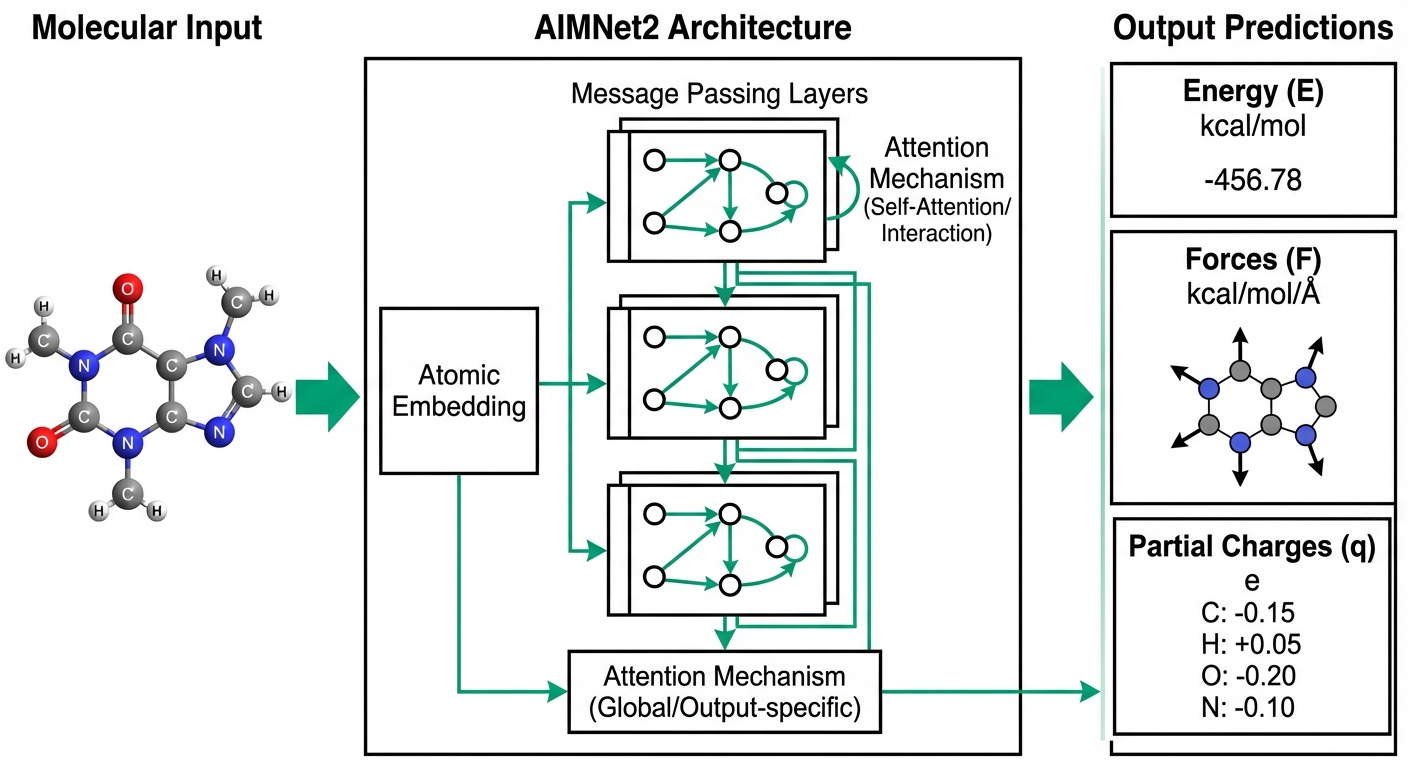
This work culminated in AIMNet2, a transferable neural network potential capable of treating neutral molecules, ions, radicals, and reactive intermediates within a unified computational framework. AIMNet2 was explicitly designed to avoid the narrow specialization that limits the utility of many machine learning potentials in exploratory settings. The model achieves competitive accuracy across diverse chemical datasets—encompassing 14 elements (H, B, C, N, O, F, Si, P, S, Cl, As, Se, Br, I) that cover more than 90% of drug-like molecules—while maintaining computational efficiency that enables high-throughput applications. Training on approximately 20 million hybrid DFT calculations using range-separated functionals ensures broad coverage of conformational and electronic diversity. The architecture combines ML-parameterized short-range interactions with physics-based long-range electrostatics through flexible Coulomb treatments including damped shifted force (DSF) and Ewald summation methods. Integration with standard simulation packages including ASE and PySisyphus enables seamless deployment in molecular dynamics, geometry optimization, and reaction pathway workflows.
Extensions of this framework include AIMNet2-NSE, which enables modeling of open-shell and spin-polarized chemistry essential for treating radicals, triplet states, and systems exhibiting significant spin contamination. These developments demonstrate that chemical transferability can be achieved through careful attention to representation design, scalable neural network architectures, and training strategies informed by physical intuition. The original ANI publication has accumulated over 1,500 citations, and the methods are now deployed in production environments at major pharmaceutical and chemical companies including Dow Chemical, BASF, GSK, Pfizer, and Genentech, validating their practical utility beyond academic benchmarks.
Reaction Modeling at Scale
A major theme of my research is the application of machine learning potentials to chemical reactivity and reaction network exploration. Reactive processes demand accurate forces and smooth potential energy surfaces across bond-breaking and bond-forming events—regimes where traditional force fields fundamentally fail and quantum chemistry becomes computationally prohibitive for extensive pathway exploration.
I addressed this challenge through the development of AIMNet2-rxn, a machine-learned potential trained on millions of reaction pathways that enables generalized reaction modeling at unprecedented scale while retaining near-quantum chemical accuracy for transition state energetics. This capability supports mechanistic insight into complex multi-step transformations, systematic reaction discovery across large chemical spaces, and large-scale screening without sacrificing the reliability required for chemical decision-making. Related work has demonstrated the applicability of transferable ML potentials to transition-metal catalysis, including palladium-catalyzed cross-coupling reactions where accurate treatment of reactive organometallic intermediates is essential for predicting regioselectivity and understanding catalyst deactivation pathways.
Methodological Principles
Several methodological principles unify my research on machine learning potentials. Chemical transferability serves as a primary design objective from the outset, rather than a property to be assessed after model development. Physically informed representations ensure consistency with known symmetries, scaling laws, and the locality of chemical bonding. Robust behavior outside the training distribution receives particular attention, especially for reactive and charged systems where extrapolation failures can lead to qualitatively incorrect predictions. Practical integration with molecular dynamics engines, reaction discovery pipelines, and automated experimentation workflows ensures that methodological advances translate into deployable tools.
Key innovations across this body of work include the demonstration that ML potentials trained on small molecules can transfer effectively to larger systems through appropriate representation design; active learning protocols that automate the expansion of training datasets by identifying regions of chemical space where model uncertainty is highest; extension to ionic species and charged molecular fragments through the AIMNet2 framework; incorporation of long-range physics including electrostatics and dispersion corrections; and reactive potential development enabling exploration of chemical transformations in condensed phases.
I have emphasized the importance of critical evaluation and rigorous benchmarking of machine learning interatomic potentials, particularly as these models are increasingly deployed in settings beyond their original training domains. The development of standardized benchmarks and transparent reporting of model limitations is essential for responsible deployment in discovery applications.
Outlook and Future Directions
Looking forward, my research aims to expand the scope and reliability of machine learning potentials in several directions. Unified potentials that seamlessly treat closed-shell, open-shell, and electronically complex systems will broaden applicability to photochemistry, catalysis, and materials under extreme conditions. Active and adaptive learning protocols, where ML potentials guide the generation of new quantum chemical reference data during simulations, will enable continuous model improvement in production workflows. Uncertainty-aware ML potentials that provide calibrated confidence estimates will enable principled decision-making in automated and high-throughput discovery campaigns, flagging predictions that require higher-level validation.
More broadly, I view machine learning potentials as a foundational technology for autonomous chemical discovery, in which ML-driven simulation, generative molecular design, and quantum refinement operate in a closed loop. The long-term objective is not to replace quantum chemistry, but to extend its reach and make high-accuracy atomistic modeling routine for complex chemical problems that were previously intractable. We are advancing toward what might be termed “Chemical Intelligence”—moving machine learning beyond data modeling toward expert-validated inference systems capable of autonomous reasoning for research plan generation and experimental design.
Key Publications
Best practices in machine learning for chemistry
Nature Chemistry , 13 , 505–508 (2021)
Extending the Applicability of the ANI Deep Learning Molecular Potential to Sulfur and Halogens
Journal of Chemical Theory and Computation , 16 , 4192–4202 (2020)
The ANI-1ccx and ANI-1x data sets, coupled-cluster and density functional theory properties for molecules
Scientific Data , 7 (2020)
Less is more: Sampling chemical space with active learning
The Journal of Chemical Physics , 148 (2018)
ANI-1: an extensible neural network potential with DFT accuracy at force field computational cost
Chemical Science , 8 , 3192–3203 (2017)
Software & Tools
Frequently Asked Questions
What are machine learning potentials?
Machine learning potentials (MLPs) are computational models that learn the relationship between atomic positions and energies from quantum mechanical calculations. They provide near-quantum accuracy at a fraction of the computational cost, enabling molecular simulations at scales impossible with traditional ab initio methods.
What is AIMNet2 and how does it differ from other neural network potentials?
AIMNet2 is a general-purpose neural network potential trained on approximately 20 million DFT calculations spanning diverse organic chemistry. Unlike specialized potentials, AIMNet2 transfers across chemical space without retraining, predicting energies, forces, and partial charges for drug-like molecules with 1-2 kcal/mol accuracy.
How accurate are neural network potentials compared to DFT?
Modern neural network potentials like AIMNet2 achieve 1-2 kcal/mol accuracy compared to reference DFT calculations, which is sufficient for most thermochemistry and conformational analysis applications. For higher accuracy requirements, delta-learning approaches can target coupled-cluster accuracy.
Can machine learning potentials handle reactive chemistry?
Yes, specialized reactive MLPs like AIMNet2-rxn are trained on transition state regions and reaction coordinates, enabling accurate modeling of bond breaking and formation. These models achieve roughly 6 orders of magnitude speedup over DFT while maintaining chemical accuracy for reaction barriers.