De Novo Molecular Design with Deep Learning
Generative AI in Chemistry
Comprehensive research on generative models—VAEs, GANs, flow, and diffusion models—for de novo molecular design and chemical discovery.
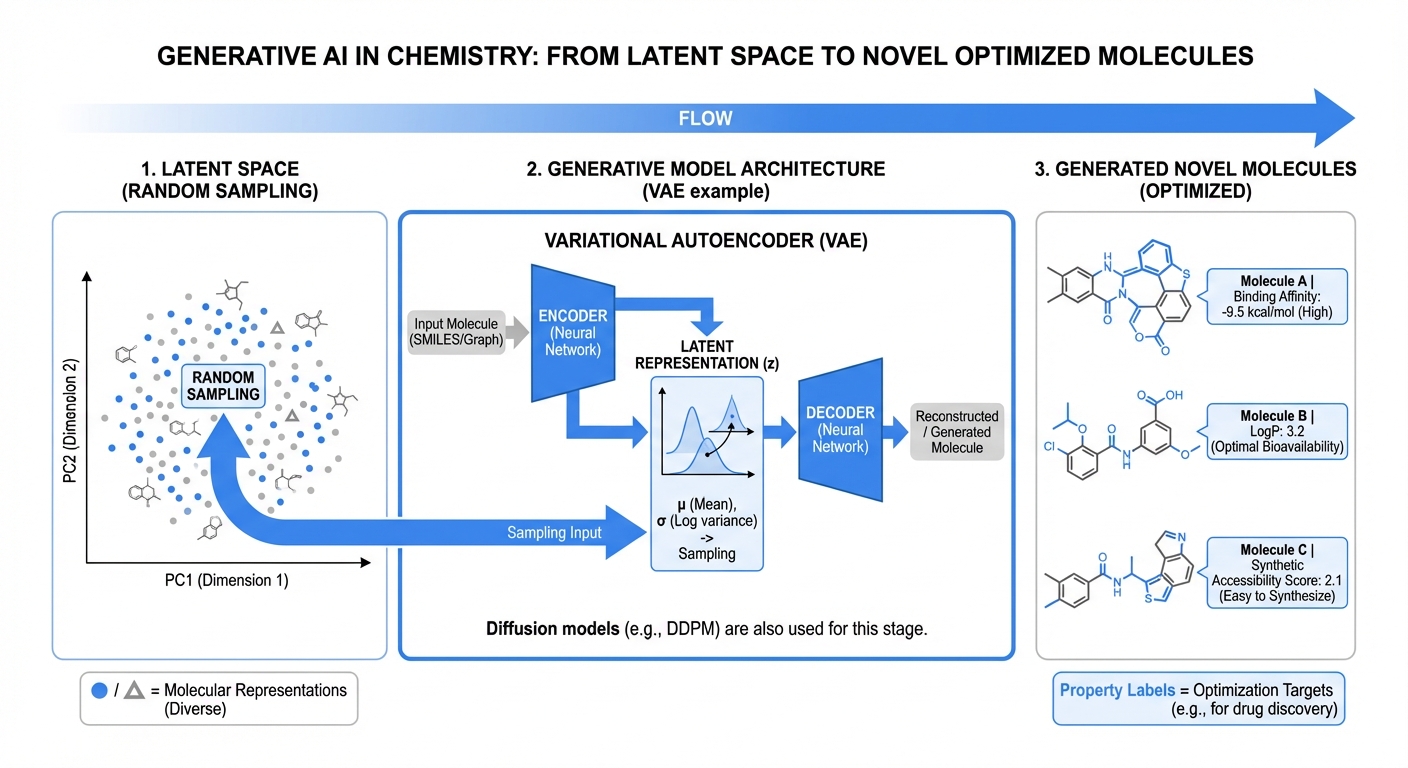
My research in generative artificial intelligence for drug discovery focuses on developing models that enable systematic exploration of chemical space under biological, physicochemical, and synthetic constraints. Drug discovery is inherently a multi-objective optimization problem, requiring simultaneous consideration of potency, selectivity, pharmacokinetics, toxicity, and synthetic accessibility. Generative AI provides a principled framework for navigating this complex design landscape by learning probability distributions over chemically valid molecules and proposing candidates that satisfy multiple competing objectives. Unlike traditional virtual screening approaches that evaluate fixed compound libraries, generative models enable de novo molecular design—the creation of entirely novel chemical entities optimized for specific applications rather than selected from existing collections.
Generative Molecular Design: From Strings to Geometries
Generative models for molecular design have undergone rapid evolution from string-based representations to increasingly sophisticated graph- and geometry-aware architectures. My foundational work with ReLeaSE (Reinforcement Learning for Structural Evolution), published in Science Advances in 2018 with over 1,000 citations, established a computational strategy integrating deep learning with reinforcement learning for de novo design. The methodology addresses the fundamental challenge of navigating chemical space to identify molecules satisfying multiple competing objectives.
The ReLeaSE architecture employs a two-phase training paradigm. In the supervised pretraining phase, a stack-augmented recurrent neural network learns to produce chemically valid SMILES strings while a separate predictive network learns structure-property relationships. In the subsequent reinforcement learning phase, both networks are trained jointly using policy gradient methods, biasing the generator toward molecules with desired biological and physicochemical properties.
The choice of stack-augmented memory networks is theoretically justified by formal language theory: valid SMILES strings constitute a context-free language requiring proper tracking of nested dependencies including bracket matching, ring annotations, and branching notation. This architecture achieves 95% validity rates compared to 86% for standard recurrent networks without stack memory.
Subsequent developments in my group have explored transfer learning approaches for conditional molecular generation with continuous property targets. The RRCGAN (Regressional Reinforcement Conditional GAN) framework, published in Digital Discovery, addresses two critical challenges: generating molecules with specific user-defined property values rather than binary classifications, and discovering molecules with properties beyond the range present in training data. Through iterative transfer learning protocols, the system progressively expands the accessible property envelope, demonstrating extrapolation to regimes absent from original training distributions. Experimental validation through density functional theory calculations confirms the physical plausibility of generated structures.
Property Prediction and Multi-Objective Optimization
Generation alone is insufficient for drug discovery applications. Generated molecules must be rigorously evaluated with respect to biological activity, selectivity, ADMET properties, and synthetic feasibility before experimental validation. Generative AI is therefore most effective when embedded within closed-loop workflows that tightly couple generation with predictive models for property estimation.
Machine learning models for molecular property prediction have demonstrated strong performance across a wide range of drug-relevant endpoints including binding affinity, metabolic stability, and toxicity. In my research program, generative models are coupled to these predictive engines to enable conditional generation and optimization, where molecular proposals are guided by learned structure-property relationships rather than exhaustive enumeration. This integrated approach enables multi-objective optimization that balances potency, selectivity, and ADMET properties simultaneously; efficient exploration of chemical space beyond the bounds of human intuition; substantial reduction in the number of compounds requiring experimental validation; and accelerated discovery timelines from initial concept to viable drug candidates.
Feedback Loops and Active Learning
A defining feature of modern generative drug discovery pipelines is the use of feedback loops, in which information from predictive models, physics-based simulations, or experimental assays is returned to improve both the generative and predictive components. This paradigm enables adaptive exploration and efficient allocation of computational and experimental resources—critical considerations given the cost of synthesis and biological testing.
Active learning and Bayesian optimization frameworks provide a natural foundation for such closed-loop systems. By iteratively proposing candidates, evaluating them through computation or experiment, and updating models based on outcomes, these workflows accelerate convergence toward high-quality drug candidates while reducing the number of compounds that must be made and tested. My group’s work on validated EGFR inhibitors, published in Communications Chemistry, demonstrated this approach by experimentally confirming the activity of computationally designed compounds generated through deep reinforcement learning. The study introduced solutions to the sparse reward problem inherent in reinforcement learning for molecular design—where the majority of generated molecules are predicted as inactive—through modified reward shaping, temperature-based sampling strategies, and experience replay mechanisms.
A key insight from this work is that the bottleneck in generative molecular design is often not generation itself, but the generalization of property predictors to novel chemical space. Active learning addresses this limitation by strategically selecting molecules that maximally improve model coverage, enabling more reliable predictions for subsequent generations.
Integration with Physics-Based Models
While many generative drug discovery approaches rely primarily on ligand-based or data-driven scoring functions, my research emphasizes the integration of generative AI with physics-based and physically informed models, including machine learning interatomic potentials and quantum chemistry. This hybrid approach enables evaluation of molecular stability through conformational energetics, assessment of reactivity and metabolic susceptibility through reaction modeling, validation of binding modes through molecular dynamics simulations, and quantification of strain energy and intramolecular interactions that influence binding.
Such physics-informed approaches are increasingly recognized as essential for improving robustness and extrapolation in molecular design. By combining generative AI with physically grounded evaluation, it becomes possible to filter unrealistic candidates early in the design process and focus experimental validation efforts on molecules that are both predicted active and chemically viable. My perspective article in JACS, “Generative Models as an Emerging Paradigm in the Chemical Sciences,” synthesizes the broader landscape of this field, contrasting discriminative models that learn to predict properties given structures with generative models that exploit joint probability distributions to enable inverse design—starting from target property profiles and optimizing molecular structure rather than screening through combinatorial libraries.
Outlook and Future Directions
Looking forward, my research aims to advance generative AI for drug discovery through geometry- and conformation-aware generation that enables tighter integration with structure-based drug design and explicit consideration of three-dimensional binding interactions; uncertainty-aware generative models that quantify confidence in predictions and guide experimental validation toward compounds with the highest expected information value; and autonomous discovery systems that seamlessly integrate generation, prediction, simulation, and experimentation in closed-loop workflows requiring minimal human intervention.
The paradigm is fundamentally transforming how chemists approach molecular design—from screening existing compound collections to generating entirely new molecular entities optimized for specific therapeutic applications. When combined with predictive modeling, physical simulation, and active learning, generative models offer a scalable and scientifically grounded approach to exploring chemical space and accelerating therapeutic innovation. The long-term vision is a future where the design-make-test-analyze cycle is substantially automated, with generative AI proposing candidates, robotic synthesis producing them, and automated assays evaluating their properties—all orchestrated by adaptive algorithms that learn from each iteration.
Key Publications
Generative Models as an Emerging Paradigm in the Chemical Sciences
Journal of the American Chemical Society , 145 , 8736–8750 (2023)
The transformational role of GPU computing and deep learning in drug discovery
Nature Machine Intelligence , 4 , 211–221 (2022)
OpenChem: A Deep Learning Toolkit for Computational Chemistry and Drug Design
Journal of Chemical Information and Modeling , 61 , 7–13 (2021)
Development of Multimodal Machine Learning Potentials: Toward a Physics-Aware Artificial Intelligence
Accounts of Chemical Research , 54 , 1575–1585 (2021)
Deep reinforcement learning for de novo drug design
Science Advances , 4 (2018)
Software & Tools
Frequently Asked Questions
What is ReLeaSE and how does it enable de novo drug design?
ReLeaSE (Reinforcement Learning for Structural Evolution) is a computational strategy that integrates deep learning with reinforcement learning for de novo molecular design. Published in Science Advances with over 1,000 citations, it uses a stack-augmented recurrent neural network to generate chemically valid SMILES strings while policy gradient methods bias the generator toward molecules with desired biological and physicochemical properties, achieving 95% validity rates.
How do generative models differ from traditional virtual screening approaches?
Unlike traditional virtual screening that evaluates fixed compound libraries, generative models enable de novo molecular design—creating entirely novel chemical entities optimized for specific applications. They learn probability distributions over chemically valid molecules and propose candidates satisfying multiple competing objectives including potency, selectivity, pharmacokinetics, and synthetic accessibility.
What is the role of active learning in generative drug discovery?
Active learning addresses a key bottleneck in generative molecular design: the generalization of property predictors to novel chemical space. By strategically selecting molecules that maximally improve model coverage, active learning enables more reliable predictions for subsequent generations. Our validated EGFR inhibitor work demonstrated this by experimentally confirming activity of computationally designed compounds.
How does transfer learning enable property extrapolation in molecular generation?
The RRCGAN framework uses iterative transfer learning to generate molecules with properties beyond the training data distribution. By progressively expanding the accessible property envelope through refinement cycles, the system demonstrated extrapolation to regimes absent from original training distributions, with all generated structures validated through DFT calculations to confirm physical plausibility.