AIMNetCentral: Fast Machine-Learned Potentials for Molecular Dynamics
Introducing AIMNetCentral, our streamlined Python package for deploying AIMNet2 neural network potentials in molecular dynamics simulations with ASE and PySisyphus integration.
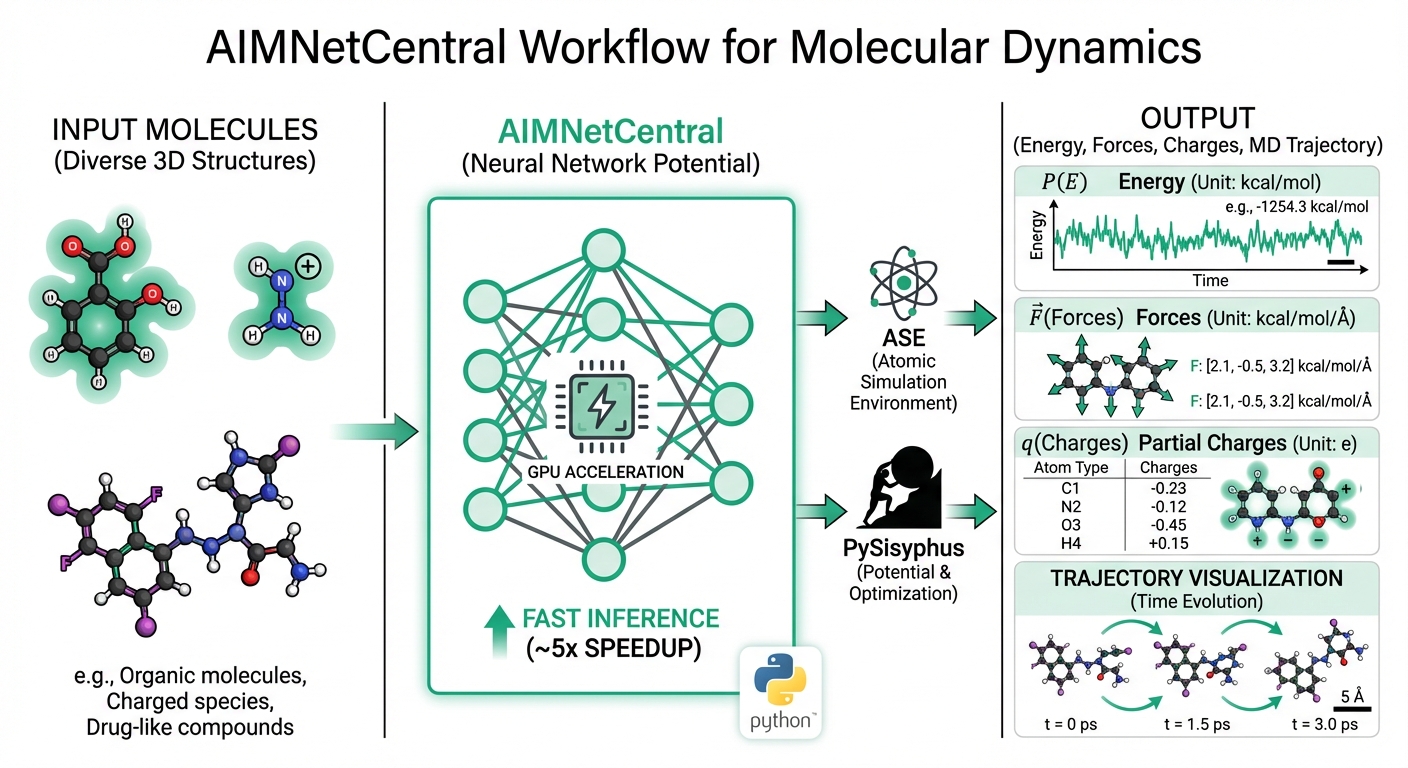
I am pleased to announce the release of AIMNetCentral, a streamlined Python package that makes it easy to deploy AIMNet2 neural network potentials in molecular dynamics simulations and geometry optimizations. This software represents our effort to lower the barrier between developing machine learning potentials and using them in practical research workflows.
AIMNetCentral serves as the central hub for running AIMNet2 models in production settings. While the scientific foundation of AIMNet2 is described in our Chemical Science paper, this package focuses on providing a clean, efficient interface for researchers who want to use these potentials without diving into the underlying implementation details. The goal is simple: install the package, load a model, and start running simulations.
The package provides seamless integration with two of the most popular frameworks in computational chemistry. For users of the Atomic Simulation Environment (ASE), AIMNetCentral offers a calculator interface that drops directly into existing workflows for geometry optimization, molecular dynamics, and property calculations. For reaction pathway studies, the package integrates with PySisyphus, enabling transition state searches and minimum energy path calculations with AIMNet2 accuracy at a fraction of the cost of density functional theory.
One of the key features I want to highlight is the flexibility in handling electrostatics. AIMNetCentral supports both Damped-Shifted Force (DSF) and Ewald summation Coulomb models, making it suitable for both molecular systems and periodic calculations. This means researchers can use the same potential for gas-phase conformational analysis and condensed-phase simulations without switching tools.
The package ships with several pre-trained model variants to address different chemical domains. The default model trained on the wB97M-D3 functional covers neutral and charged organic molecules across 14 elements. We also provide models trained on the B97-3c functional for users who prefer that level of theory, as well as specialized models for open-shell chemistry and palladium-containing systems relevant to catalysis research.
Performance has been a major focus. On GPU hardware with CUDA, enabling compiled mode provides approximately 5x speedup for molecular dynamics trajectories compared to standard inference. The calculator returns energies, forces, and atomic partial charges, with optional support for Hessian matrices and stress tensors when needed for periodic systems or vibrational analysis.
Installation requires Python 3.11 or 3.12 and can be done directly via pip from our GitHub repository. The package works on CPU out of the box, with GPU acceleration available for users with CUDA-enabled PyTorch installations.
To get started with AIMNetCentral:
pip install git+https://github.com/isayevlab/aimnetcentralI encourage researchers to explore the GitHub repository for documentation, examples, and tutorials. We welcome feedback and contributions from the community as we continue to develop tools that make machine learning potentials accessible to the broader computational chemistry community.