AIMNet2: A Neural Network Potential for Neutral, Charged, and Element-Organic Molecules
Our AIMNet2 paper is now published in Chemical Science, introducing a transferable neural network potential trained on 20 million DFT calculations that covers 14 chemical elements.
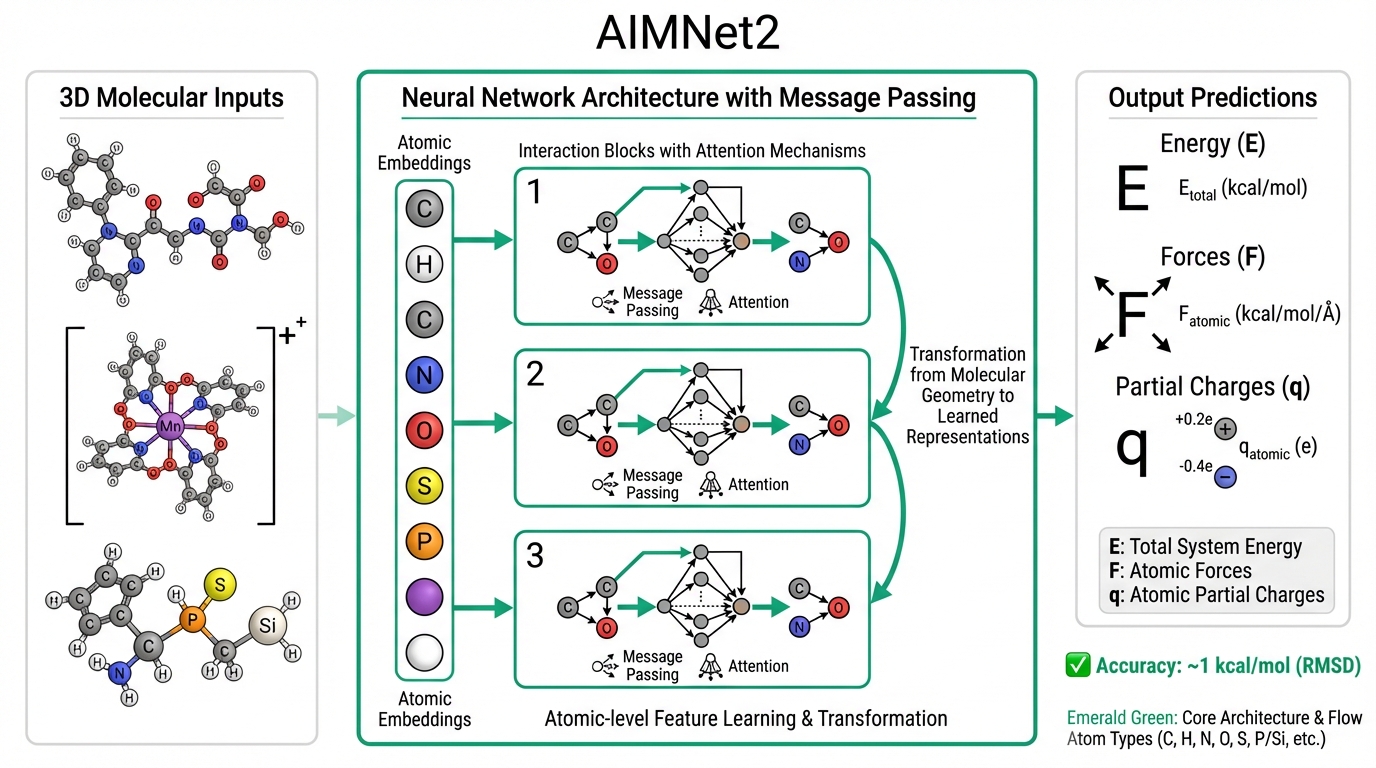
We are pleased to announce the publication of our work on AIMNet2 in Chemical Science. This paper represents a significant milestone in machine learning interatomic potentials, demonstrating that a single neural network model can achieve broad chemical transferability without the need for application-specific retraining.
Machine learning potentials have transformed computational chemistry by offering quantum mechanical accuracy at a fraction of the computational cost. However, most existing models require users to curate training data and retrain for each new chemical system—a substantial barrier to widespread adoption. AIMNet2 addresses this limitation directly by providing a general-purpose potential that works out of the box across diverse organic and element-organic chemistry.
The model was trained on approximately 20 million hybrid density functional theory calculations spanning 14 chemical elements: hydrogen, boron, carbon, nitrogen, oxygen, fluorine, silicon, phosphorus, sulfur, chlorine, arsenic, selenium, bromine, and iodine. This element coverage encompasses more than 90% of drug-like molecules and a broad range of organometallic and element-organic compounds. Critically, AIMNet2 handles both neutral molecules and charged species—including anions, cations, and zwitterions—within a unified framework, eliminating the need for separate models for different charge states.
The architecture combines ML-parameterized short-range interactions with physics-based long-range electrostatics, ensuring physically meaningful behavior across different molecular environments. The model predicts not only energies and forces but also atomic partial charges, enabling accurate treatment of electrostatic interactions in condensed-phase simulations and providing chemically interpretable outputs.
Extensive benchmarking demonstrates that AIMNet2 achieves performance comparable to DFT across multiple challenging tasks. For conformational energies and geometries, the model reproduces DFT results with mean absolute errors below 1 kcal/mol. Torsion energy profiles, which are critical for understanding molecular flexibility, show excellent agreement with reference calculations. The model also performs well on interaction energies for non-covalent complexes and maintains accuracy for geometry optimization of diverse molecular structures.
Perhaps most importantly, AIMNet2 demonstrates strong transferability to chemical systems outside its explicit training distribution. This generalization capability means researchers can apply the model to novel compounds with confidence, knowing that performance will remain reliable even for molecules the model has never seen during training.
The practical impact of AIMNet2 extends across multiple domains. In drug discovery, the model enables rapid evaluation of conformational landscapes, binding pose assessment, and property prediction for large compound libraries. For reaction modeling, AIMNet2 provides the accuracy needed to characterize transition states and reaction pathways at computational costs that permit exploration of complex reaction networks. Materials scientists can use the model to screen candidate compounds and predict properties relevant to molecular crystals, polymers, and organic electronics.
AIMNet2 is fully open source and available through our GitHub repository. You can use it as a PyTorch-based interatomic potential in your own workflows:
import torch
from aimnet.calculators import AIMNet2Calculator
# Load the AIMNet2 model (e.g., WB97M-V ensemble)
calc = AIMNet2Calculator("aimnet2_wb97m-v_ens")
# The calculator can be used directly with ASE for simulations:
# atoms.set_calculator(calc)
# energy = atoms.get_potential_energy()Installation is straightforward via pip, and the model integrates seamlessly with popular simulation packages including ASE and PySisyphus. We have also provided tutorials and example workflows to help new users get started quickly.
Read the full paper: AIMNet2: a neural network potential to meet your neutral, charged, organic, and elemental-organic needs — Dylan M. Anstine, Roman Zubatyuk, and Olexandr Isayev, Chemical Science, 2025, 16, 10.1039/D4SC08572H.
We hope AIMNet2 will serve as a valuable tool for the computational chemistry community, lowering barriers to accurate molecular simulation and accelerating discovery across chemistry and the life sciences.