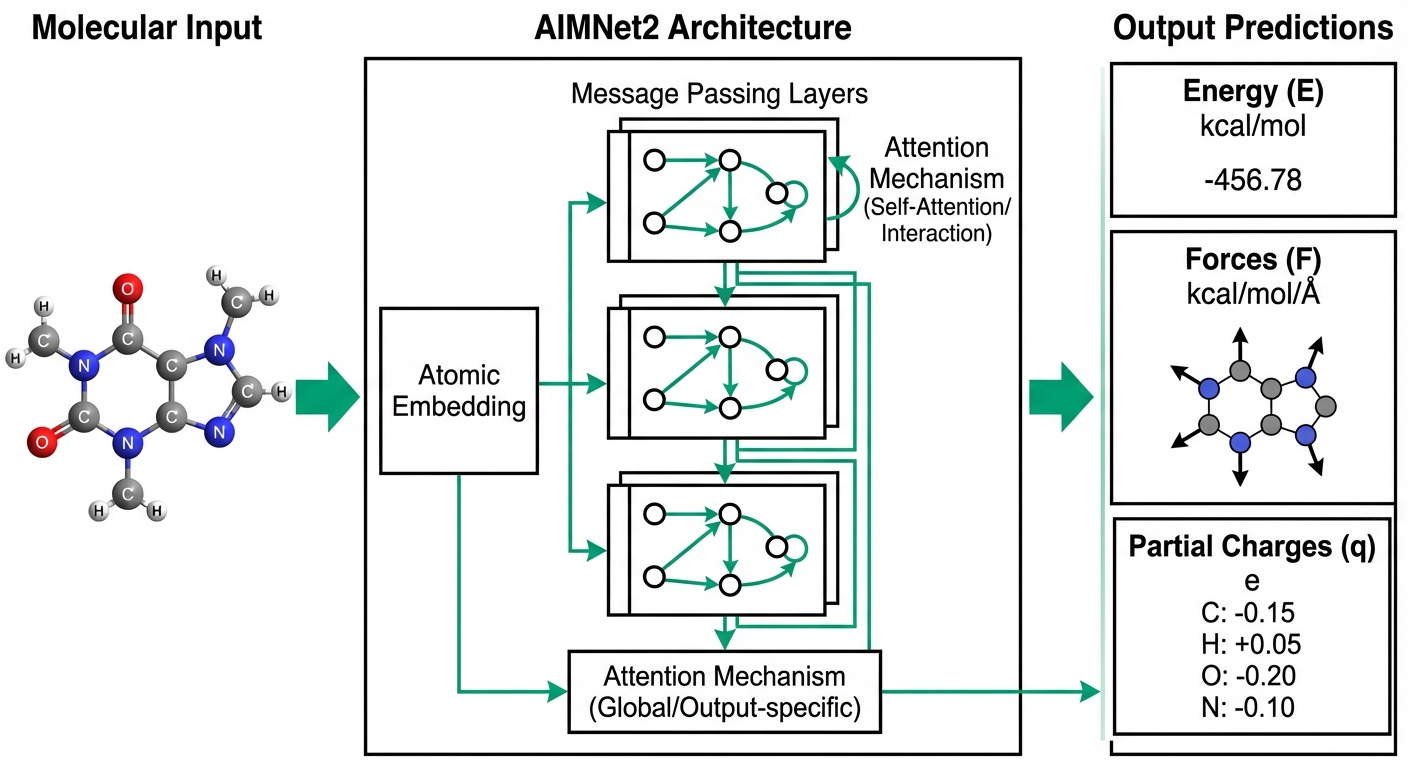
Machine Learning Potentials
Learning Quantum Mechanics with Neural Networks
Transferable neural network potentials bridging quantum accuracy with computational efficiency
Explore research →Carnegie Mellon University · Department of Chemistry
I am the Carl and Amy Jones Professor of Chemistry at Carnegie Mellon University. My research focuses on solving fundamental chemical problems through machine learning, molecular modeling, and quantum mechanics.
Since 2016, my work has pioneered research at the interface between ML and quantum chemistry, resulting in several families of atomistic machine learning potentials now used by leading laboratories and companies worldwide.


Please support Ukraine! You can help by donating to the numerous organizations. Russia must be defeated and punished for the war crimes.
Learning Quantum Mechanics with Neural Networks
Transferable neural network potentials bridging quantum accuracy with computational efficiency
Explore research →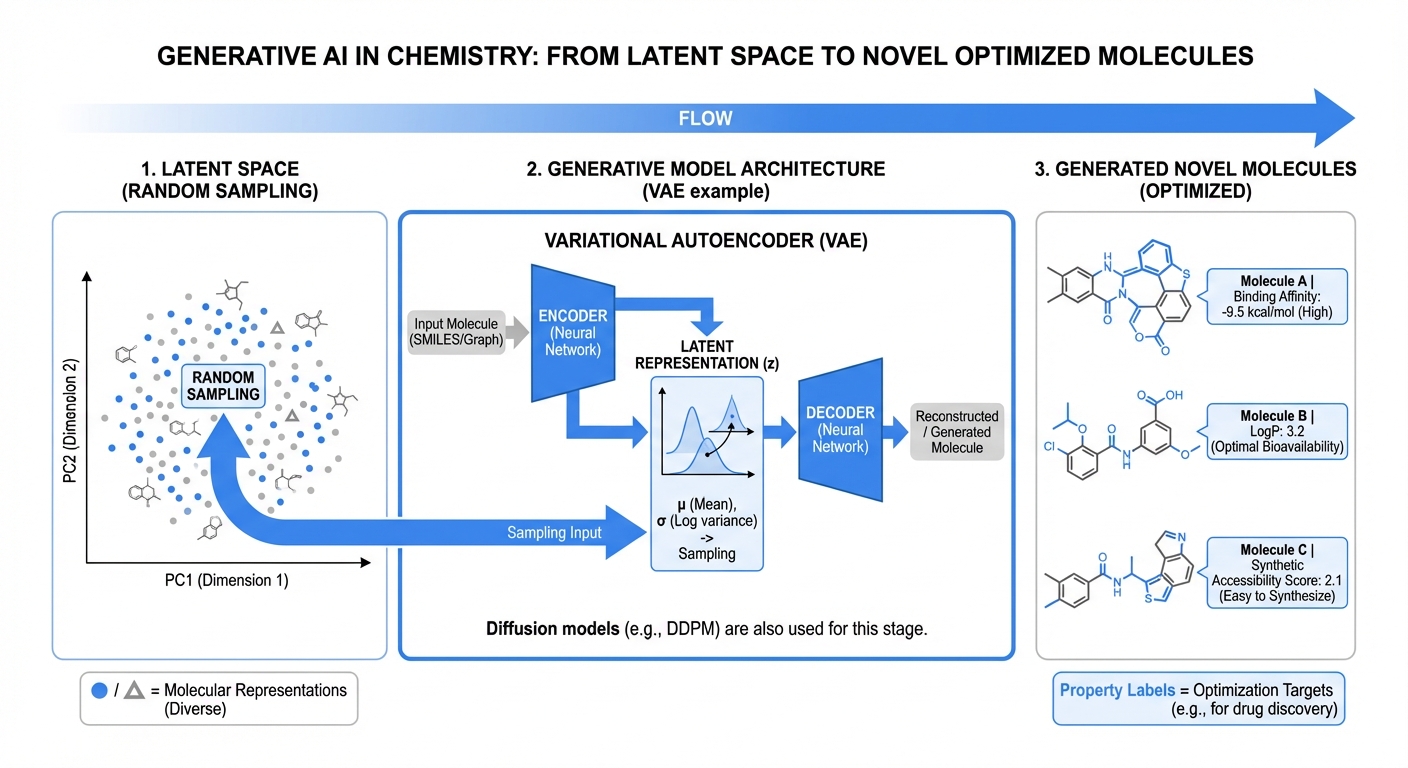
De Novo Molecular Design with Deep Learning
Generative models enabling systematic exploration of chemical space under biological and synthetic constraints
Explore research →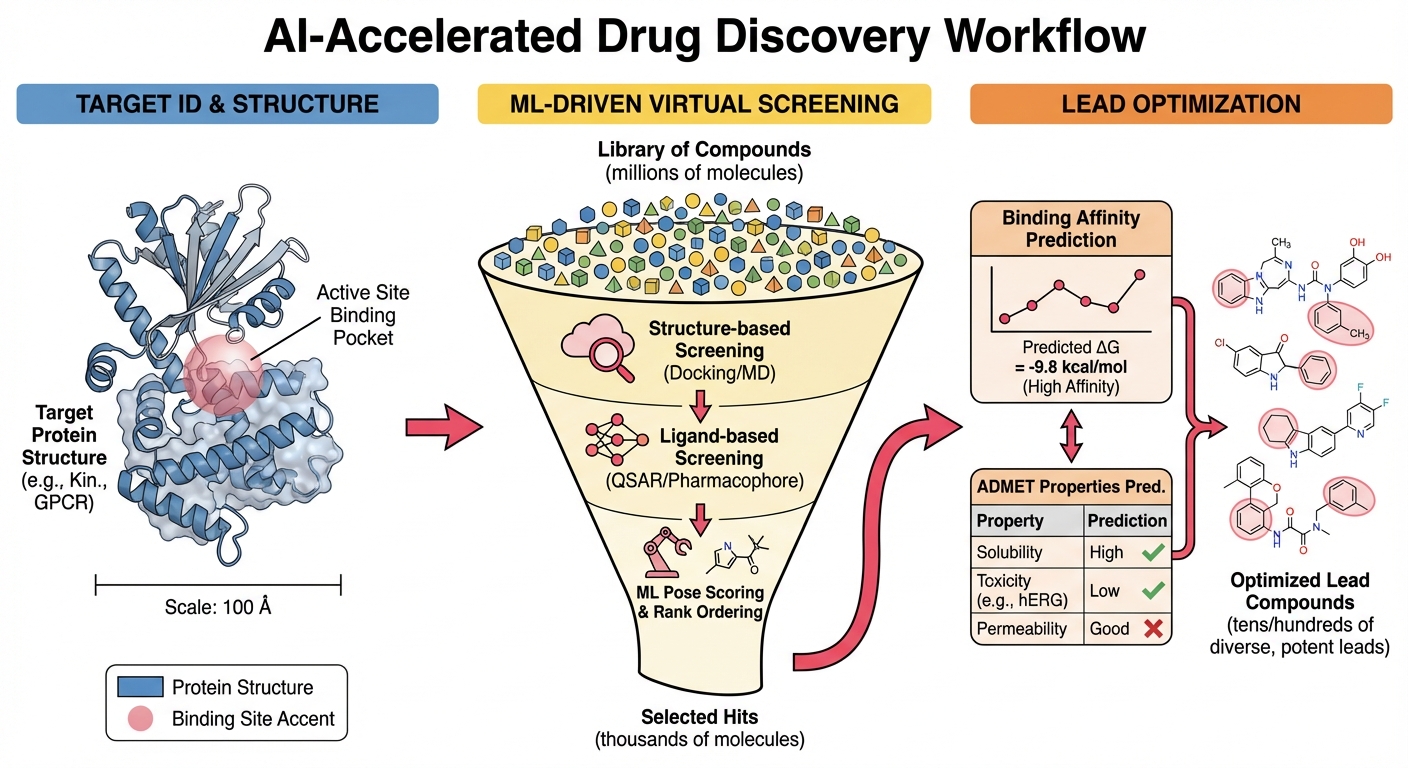
Energy-Driven Computational Methods for Medicinal Chemistry
Energy-based ML methods for reliable compound prioritization in industrial drug discovery
Explore research →Introducing AIMNetCentral, our streamlined Python package for deploying AIMNet2 neural network potentials in molecular dynamics simulations with ASE and PySisyphus integration.
Our collaborative work on AQuaRef is now published in Nature Communications. This AI-enabled quantum refinement method leverages AIMNet2 to achieve unprecedented accuracy in protein structure determination from cryo-EM and X-ray crystallography data.
Our AIMNet2 paper is now published in Chemical Science, introducing a transferable neural network potential trained on 20 million DFT calculations that covers 14 chemical elements.
Journal of Chemical Theory and Computation , 22 , 2232–2242 (2026)
Digital Discovery , 5 , 754–768 (2026)
Chemical Science , 16 , 10228–10244 (2025)
Journal of Chemical Information and Modeling , 65 , 10239–10252 (2025)
Journal of Chemical Theory and Computation , 21 , 10362–10372 (2025)